Polymers for Electronic Applications pdf pdf
Teks penuh
Gambar
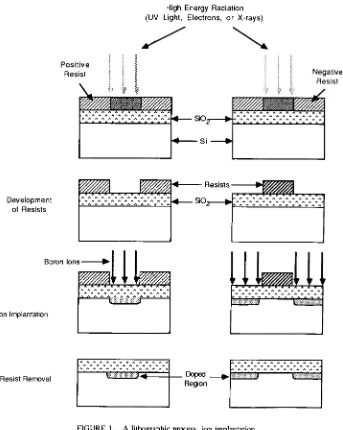
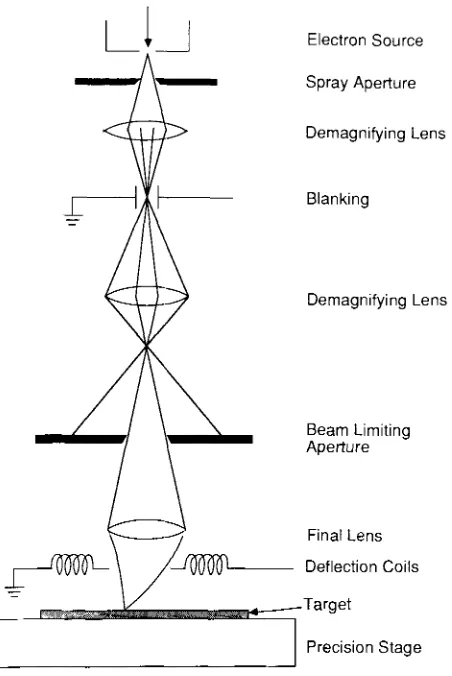
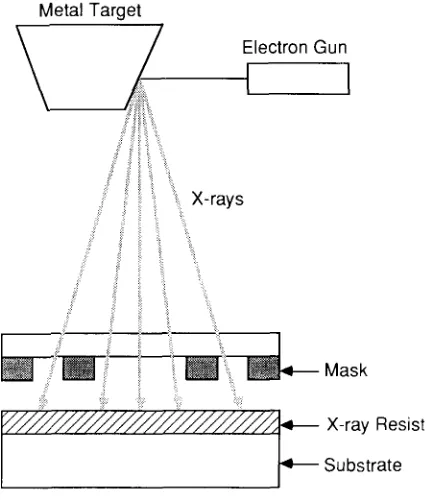
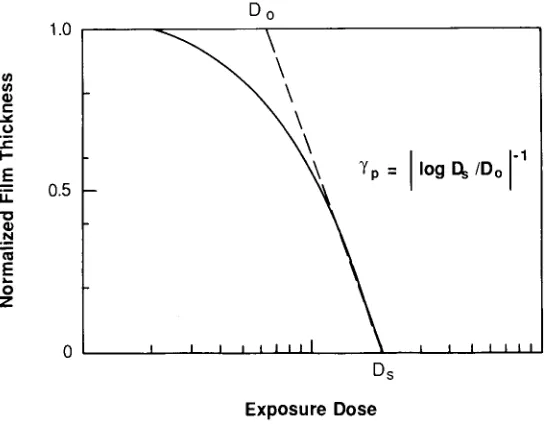

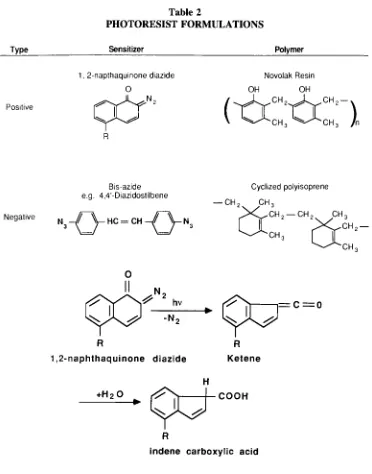


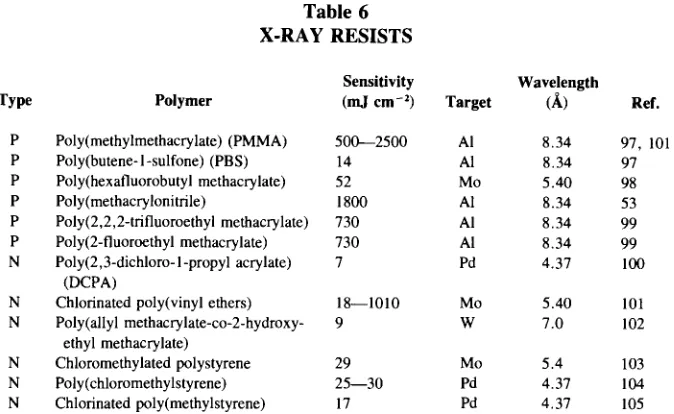
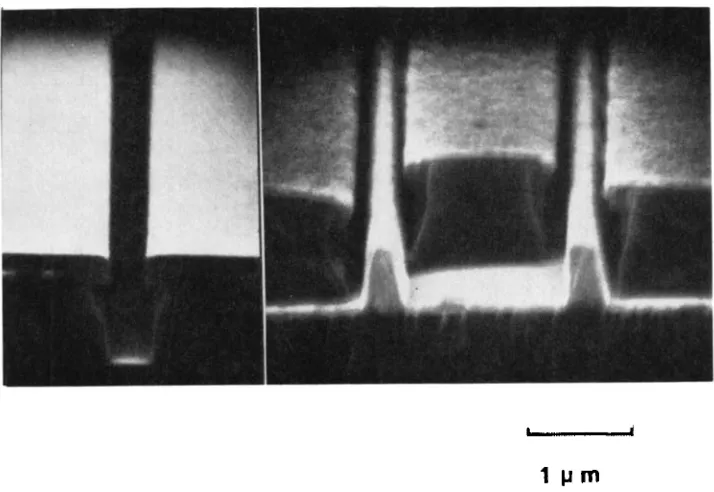
Garis besar
Dokumen terkait
[r]
An Introduction to the Syaria Economic Mohamad Hidayat Zikrul Hakim Jakarta 297.63 Ekonomi Syari'ah 2010 Indonesia 1 Bank Syari'ah dari teori ke praktik Muhammad Syafi'I Antonio
- Gap , didefinisikan sebagai waktu / jarak antara kendaraan pada arus mayor (utama) yang dipertimbangkan oleh pengemudi pada arus minor yang berharap untuk bergabung ke dalam
The higher loss in pigment composition, at 30 and 37¡C, was observed during the Ðrst month, while jams stored at 20¡C showed a progressive decrease of anthocyanins during the Ðrst
Lima besar prioritas yang diprioritaskan untuk diperbaiki adalah memiliki model transportasi yang berbeda untuk pengiriman produk ke pelanggan ( Delivery System ), secara teknis
Penelitian ini secara khusus akan membuat skema autentikasi berbasis kunci yang dipakai bersama dengan model pertukaran kunci Diffie-Hellman berdasarkan teori matriks invers..
terhadap surat perjanjian ini maka kami tutup dan izin usaha kami siap untuk dicabut. Surat pemyataan ini kami buat dengan sebenamya dalam keadaan sadar tanpa
bahwa berdasarkan pertimbangan sebagaimana dimaksud dalam huruf a, perlu menetapkan Keputusan Bupati tentang Pembentukan Tim Pengelolaan Gaji Pegawai
