Validasi metode penetapan kadar kurkumin dalam sediaan cair obat herbal terstandar merk Kiranti secara kromatografi cair kinerja tinggi fase terbalik - USD Repository
Teks penuh
Gambar
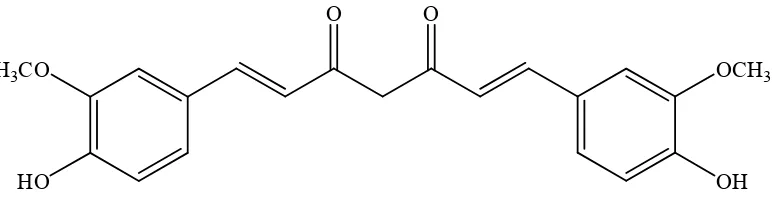

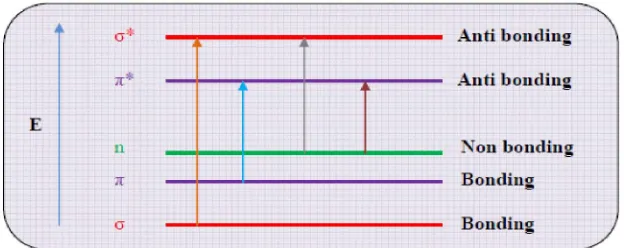

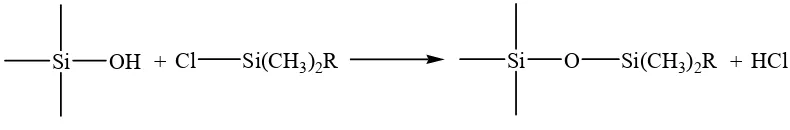
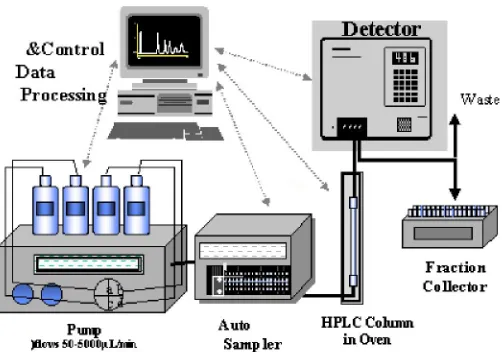
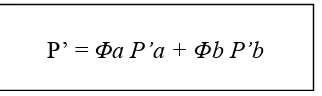
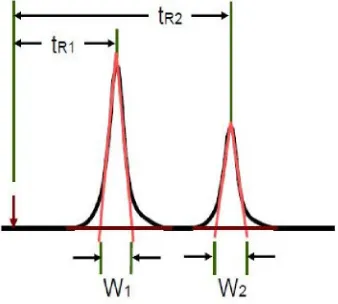
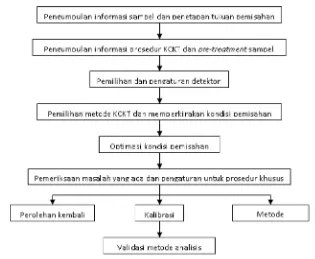

Dokumen terkait
Berdasarkan data dari Puskesmas Pagaden Barat pemberian ASI Eksklusif yang paling rendah yaitu Di Desa Cidadap sebanyak 51% dari 110 bayi.Tujuan dari penelitian
Penerbitan jurnal membutuhkan pengelolaan data yang baik dan dapat dengan mudah digunakan oleh pengguna (reviewer dan redaksi), hal ini dapat diwujudkan dengan penggunaan Sistem
Ada juga pengunjung yang belum terdaftar memasuki ruang perpustakaan yang berakibat terlalu bebasnya memasuki perpustakaan dan semakin besarnya peluang buku yang hilang
Pajak penghasilan terkait pos-pos yang akan direklasifikasi ke laba rugi (342,748) PENGHASILAN KOMPREHENSIF LAIN TAHUN BERJALAN - NET PAJAK PENGHASILAN TERKAIT 26,481,074..
Apabila pemerintah kabupaten tidak melakukan penyesuaian kembali maka peraturan daerah yang mengatur tentang retribusi akan batal demi hukum berdasarkan ketentuan Pasal
Pembagian daerah Indonesia dalam daerah-daerah besar dan kecil, dengan bentuk susunan pemerintahannya ditetapkan dengan undang-undang, dengan mengingat dasar permusyawaratan
Universitas Bina Darma memastikan bahwa apabila ditemukan ketidaksesuaian pada proses belajar mengajar, mahasiswa (tidak lulus ujian), hasil penelitian dan
Upaya apakah yang dilakukan untuk mengatasi kendala-kendala yang dihadapi oleh SMK Kristen Salatiga dalam melaksanakan Kurikulum Tingkat Satuan Pendidikan (KTSP)?. Untuk
