Validasi metode kromatografi cair kinerja tinggi fase terbalik untuk penetapan kadar salbutamol sulfat dan guaifenesin dalam sediaan sirup merek ``x`` - USD Repository
Teks penuh
Gambar



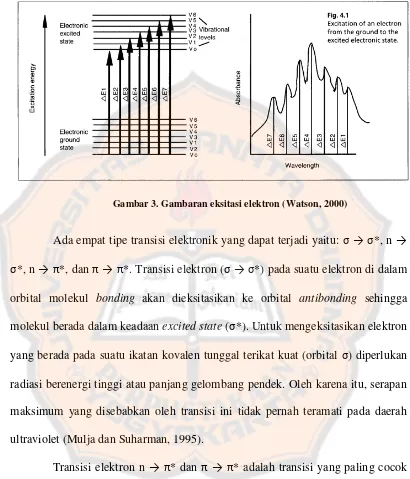

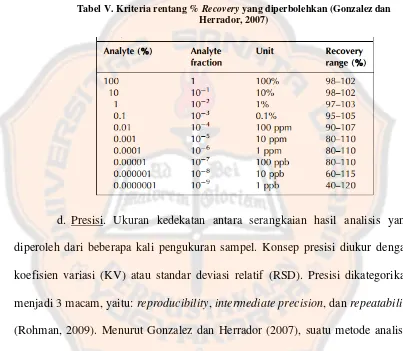
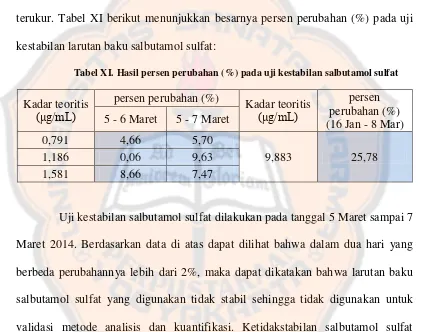
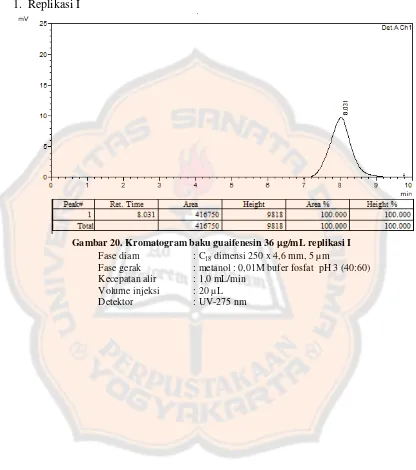
Garis besar
Dokumen terkait
Penetapan kadar parasetamol dalam sediaan sirup berdasarkan Farmakope Indonesia Edisi V, dapat ditetapkan dengan menggunakan metode Kromatografi Cair Kinerja Tinggi (KCKT)
VALIDASI METODE KROMATOGRAFI CAIR KINERJA TINGGI (KCKT) FASE TERBALIK PADA PENETAPAN KADAR NIKOTINi. DALAM EKSTRAK TEMBAKAU PADA ROKOK
=9791,15 M -1 .cm -1 , sehingga sistem KCKT fase terbalik dengan detektor UV dapat diaplikasikan untuk analisis kadar asam askorbat dalam larutan injeksi obat. pemutih
Penetapan kadar nikotin di dalam ekstrak etanolik daun tembakau dapat dilakukan dengan menggunakan metode kromatografi cair kinerja tinggi (KCKT) fase terbalik..
Sejauh penelusuran pustaka yang telah dilakukan oleh peneliti, penelitian tentang validasi metode kromatografi cair kinerja tinggi fase terbalik pada penetapan kadar
Widjaja, M., 2011, Validasi Metode Penetapan Kadar Kurkumin dalam Sediaan Cair Obat Herbal Terstandar Merk Kiranti Secara Kromatografi Cair Kinerja Tinggi Fase Terbalik, Skripsi
OPTIMASI KOMPOSISI DAN KECEPATAN ALIR FASE GERAK SISTEM KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK PADA PEMISAHAN SALBUTAMOL SULFAT DAN GUAIFENESIN DALAM.. SEDIAAN OBAT SI
Metode KCKT fase terbalik untuk penetapan kadar bisfenol A pada air minum dalam botol plastik dan botol air minum yang diberi pengaruh penyinaran matahari tropis
